Illumina sequencing submission
Illumina Short Read Sequencing Technology
Short-read sequencing, primarily powered by Illumina technology, is one of the most widely used methods for generating high-throughput genomic data. Illumina sequencing platforms utilize sequencing by synthesis (SBS), a method that involves the stepwise incorporation of fluorescently labeled nucleotides during DNA synthesis. This process enables massively parallel sequencing, producing millions to billions of short reads (typically 50–300 base pairs) per run.
The workflow typically involves library preparation, where DNA or RNA is fragmented and adapters are ligated to form a final library molecule (structure illustrated below).
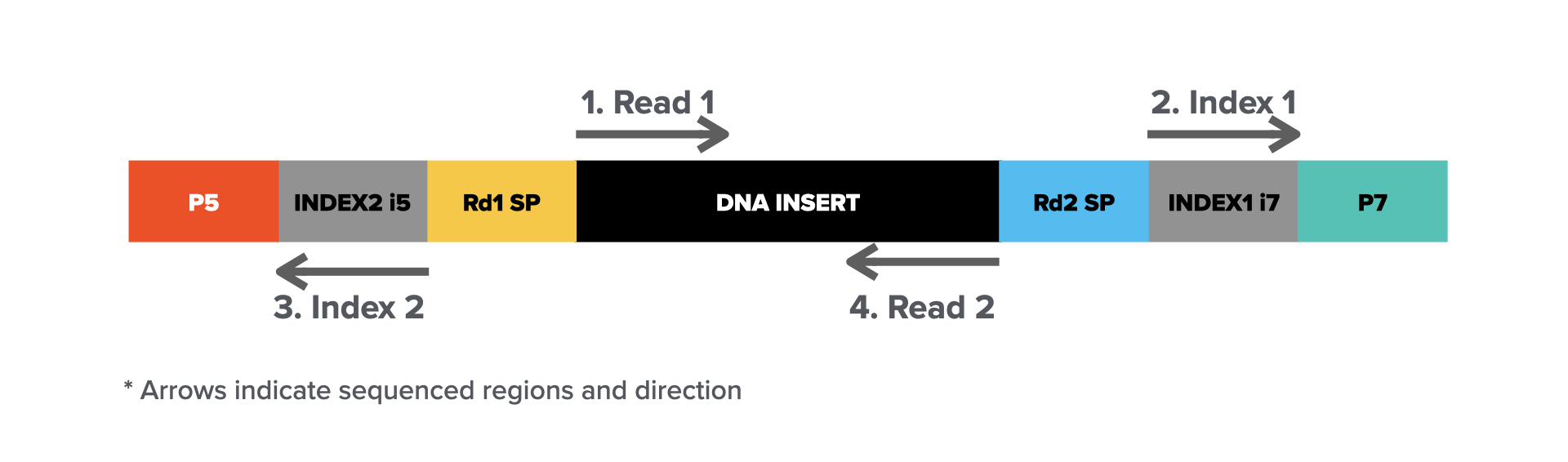
Sequencing starts by cluster amplification on a flow cell, cyclic sequencing-by-synthesis (steps illustrated above), and base calling via imaging. The high accuracy, scalability, and cost-effectiveness of Illumina sequencing make it a standard for applications such as whole-genome sequencing (WGS), RNA sequencing (RNA-seq), exome sequencing, and epigenomic studies. However, the short read length presents challenges in resolving complex genomic regions, structural variations, and long-range connectivity, which are areas where long-read sequencing technologies complement short-read approaches.
Multiplexing for High-throughput Sequencing
Multiplexing in Illumina sequencing allows multiple samples to be pooled and sequenced in a single run by incorporating unique index sequences (barcodes) into each library. These indexes are added during library preparation and later used to demultiplex reads after sequencing. Multiplexing improves efficiency, reduces costs, and enables comparative studies by maximizing data output per run. Dual-indexing helps prevent errors like index hopping, ensuring accurate sample identification. Proper library balancing and careful index selection are crucial for high-quality results in applications like RNA-seq, metagenomics, and whole-genome sequencing. If you have any questions, please contact seq-team@sf.czbiohub.org.
Resources
- Illumina Website
- Next-Generation Sequencing Basics
- Illumina Adapter Sequences
- Indexed Adapter Pooling Guide
- Sequencing Coverage Calculator
- Adapter Dimer Problem
- Removing Adapter Dimers with SPRI Beads
Sequencing submission overview
This sequencing submission system is for Biohub groups and onboarded Biohub Investigators only. If you are a new investigator, please reach out to Norma Neff with a brief description of your interests and proposed scope of work for sequencing-based experiments. We currently only accept prepared and pooled libraries. If you have questions about what library protocol to use based on your application of interest please refer to Illumina's library preparation kits or reach out to our team at seq-team@sf.czbiohub.org.
Submission instructions
Please read the current submission instructions carefully before proceeding with a submission.
Use the submission form to create an initial submission for Illumina Sequencing. Please note that in this system you will need to download, complete and upload a samplesheet as a CSV file during the initial submission process, prior to dropping off your library pool. Please refer to the instructions for sample sheet requirements.
Dropping off your library pool
Once you receive an email confirmation, you may drop off or mail your library pool to CZ Biohub SF. You will receive another email once the library has been loaded onto the sequencer (approved), and if you need to fix your sample sheet (fix sample sheet), or the data is ready for downloading (delivered). Submissions are declined automatically in the event of lack of funds, and also by our team if there was an error in the submission.
Library pool minimum volume and concentration requirements
The minimum requirements for each sequencer are listed below. Please place the pool in a 1.5 mL tube, we DO NOT accept any other format.- iSeq: ≥ 10 μL @ 4-20 nM
- MiSeq i100: ≥ 30 μL @ 4-20 nM
- NextSeq 2000: ≥ 15 μL @ 4-20 nM
- NovaSeq X 1.5B: ≥ 50 μL @ 4-20 nM (or ≥ 20 μL per lane)
- NovaSeq X 10B: ≥ 150 μL @ 4-20 nM (or ≥ 20 μL per lane)
- NovaSeq X 25B: ≥ 200 μL @ 4-20 nM (or ≥ 30 μL per lane)
After you have prepared your library pool ready to send to CZ Biohub SF Genomics Platform for sequencing, a QC step is required for submissions from CZ Biohub Chicago and CZ Biohub New York, and recommended for all other submissions. Please read the guide here.
SF Mission Bay drop off
Samples can be either dropped off (Mondays and Wednesdays 9AM-12PM only) or shipped (to arrive any weekday) to the CZ Biohub SF location in Mission Bay. Our address is:
Chan Zuckerberg Biohub SF
499 Illinois St, 4th floor
San Francisco, CA 94158
ATTN: Genomics Platform
Once you enter the lobby of 499 Illinois St., check in with personnel at the front desk, and they will lead you to the sample drop box located in the mail room on the ground floor. Scan the QR code on the box, enter your information (Name, library name, number of libraries, etc.) to register the sample drop off and place the 1.5 mL tube through the top front insert of the metal box. Please DO NOT place extra ice, or dry ice in the box.
Note: CZ Biohub SF's lab operations team and the front desk cannot accept your samples, so please follow the guidelines above set by the Genomics Platform team.
Library QC
Once we receive your library we will perform a quality check before loading the library on the sequencer. We will reach out to the submitter via email if we need further information.
Sequencing turnaround time
The turnaround time for sequencing is dependent on the number of requests in our queue. Expect a 2-3 week turnaround time from full submission (form completion and library drop-off) to data delivery, expect longer turnaround for partial flowcell requests and wrong sample sheets provided.
Data delivery
The demultiplexed data will be delivered to you via email, which contains instructions to access your data. For external collaborators, you will receive an AWS token for data download from the cloud. Please make sure that you have AWS CLI installed on your computer. The token is valid for 36 hours, so please download your data as soon as you receive an email from the Genomics Platform team containing the token.
Sample submission form
Once you have read the instructions above, please click the button below to proceed to the sample submission form.